Select The Ring Flip For The Following Compound
Onlines
Mar 16, 2025 · 6 min read
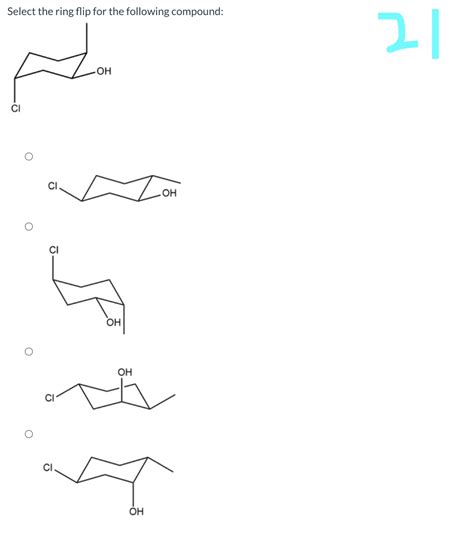
Table of Contents
Ring Flips in Cyclohexane: A Deep Dive into Conformational Analysis
The seemingly simple molecule cyclohexane presents a fascinating case study in conformational analysis. Its ability to undergo ring flips, interconverting between chair conformations, has profound implications for its reactivity and properties. This article will delve into the intricacies of cyclohexane ring flips, focusing on how to select the most stable conformation for a given substituted cyclohexane. We'll explore the 1,3-diaxial interactions that govern conformational stability, delve into the principles of A values, and examine how to predict the preferred conformation for various substitution patterns.
Understanding Cyclohexane Conformations
Cyclohexane isn't a flat, planar molecule. To minimize angle strain and torsional strain, it adopts a three-dimensional structure, primarily existing in two chair conformations. These chair conformations are interconvertible through a process called a ring flip, which involves a concerted series of bond rotations.
Chair Conformations: In a chair conformation, six carbon atoms lie in a staggered arrangement, minimizing torsional strain. The hydrogens are classified as either axial or equatorial. Axial hydrogens are oriented perpendicular to the plane of the ring, while equatorial hydrogens are roughly parallel to the plane.
Ring Flip Mechanism: The ring flip involves a transition state resembling a half-chair or twist-boat conformation. This transition state is higher in energy than the chair conformations, requiring energy input to overcome the activation barrier. Once the transition state is reached, the molecule rapidly converts to the alternative chair conformation.
Axial vs. Equatorial Positions: The key to understanding which conformation is preferred lies in the difference between axial and equatorial positions. Substituents in axial positions experience steric interactions (1,3-diaxial interactions) with axial hydrogens on the same side of the ring. These interactions destabilize the molecule, raising its energy. Substituents in equatorial positions, however, experience minimal steric interactions.
1,3-Diaxial Interactions: The Driving Force Behind Conformational Preference
1,3-diaxial interactions are the primary factor dictating the preferred conformation of substituted cyclohexanes. These are steric repulsions between a substituent in an axial position and the axial hydrogens on the carbons two carbons away (i.e., 1,3-diaxial positions). The larger the substituent, the greater the 1,3-diaxial interaction and the stronger the preference for the equatorial position.
Magnitude of 1,3-Diaxial Interactions: The strength of 1,3-diaxial interactions depends on the size of the substituent. Bulky substituents like tert-butyl groups experience very strong 1,3-diaxial interactions, almost exclusively favoring the equatorial position. Smaller substituents like methyl groups still experience significant 1,3-diaxial interactions, although the energy difference between axial and equatorial conformations is less pronounced.
A Values: Quantifying Conformational Preferences
A values provide a quantitative measure of the energy difference between the axial and equatorial conformations of a substituent. The A value is defined as the difference in free energy (ΔG°) between the axial and equatorial conformations:
A value = ΔG°(axial) - ΔG°(equatorial)
A positive A value indicates that the equatorial conformation is favored, while a negative A value indicates that the axial conformation is favored. Larger A values signify a stronger preference for the equatorial conformation. For example, the A value for a methyl group is approximately 1.7 kcal/mol, indicating a significant preference for the equatorial position.
Predicting Preferred Conformations for Mono- and Disubstituted Cyclohexanes
The principles discussed above can be applied to predict the preferred conformations for various substituted cyclohexanes.
Monosubstituted Cyclohexanes: For monosubstituted cyclohexanes, the preferred conformation is invariably the one with the substituent in the equatorial position. The larger the substituent, the more significant the preference.
Disubstituted Cyclohexanes: Disubstituted cyclohexanes present more complex scenarios. The preferred conformation depends on the relative positions and sizes of the substituents.
-
1,2-Disubstituted Cyclohexanes: These can exist as either cis or trans isomers. cis-1,2-disubstituted cyclohexanes have one equatorial and one axial substituent in their preferred conformation, while trans-1,2-disubstituted cyclohexanes have both substituents equatorial.
-
1,3-Disubstituted Cyclohexanes: Similar to 1,2-disubstituted, cis-1,3-disubstituted cyclohexanes have both substituents equatorial, whereas trans-1,3-disubstituted cyclohexanes have one equatorial and one axial substituent in the preferred conformation.
-
1,4-Disubstituted Cyclohexanes: cis-1,4-disubstituted cyclohexanes will have one axial and one equatorial substituent, while trans-1,4-disubstituted cyclohexanes will have both substituents equatorial.
In all cases, the preferred conformation minimizes the number of large substituents in the axial positions, thereby minimizing 1,3-diaxial interactions. The larger the substituents, the more pronounced this preference will be.
Selecting the Preferred Conformation: A Step-by-Step Approach
Let's outline a systematic approach for selecting the preferred conformation of a substituted cyclohexane:
-
Identify the substituents: Determine the type and size of each substituent on the cyclohexane ring.
-
Draw both chair conformations: Draw both chair conformations of the cyclohexane ring, clearly indicating the axial and equatorial positions of all substituents.
-
Assess 1,3-diaxial interactions: Identify all 1,3-diaxial interactions in both conformations. Pay close attention to the size of the substituents involved. Larger substituents will lead to stronger 1,3-diaxial interactions.
-
Compare the energies: Based on the strength of the 1,3-diaxial interactions, determine which conformation has the lower overall energy. The conformation with fewer and weaker 1,3-diaxial interactions will be the preferred conformation. Remember that the A values can guide your assessment.
-
Consider the relative positions of substituents (cis/trans): For disubstituted cyclohexanes, the cis/trans relationship dictates the possible combinations of axial and equatorial positions. This information helps refine your choice of the most stable conformation.
Examples of Ring Flip Selection
Let's consider a few specific examples to illustrate the process:
Example 1: Methylcyclohexane
Methylcyclohexane has one methyl substituent. The preferred conformation places the methyl group in the equatorial position to minimize 1,3-diaxial interactions with axial hydrogens. The axial conformation is significantly less stable.
Example 2: cis-1,2-Dimethylcyclohexane
In cis-1,2-dimethylcyclohexane, one methyl group will necessarily be axial and the other equatorial. The other chair conformation will also result in one axial and one equatorial methyl group. However, the small energy difference between these two conformations suggests they will be present in relatively equal populations at equilibrium.
Example 3: trans-1,4-Di-tert-butylcyclohexane
trans-1,4-Di-tert-butylcyclohexane presents a strong case for conformational preference. Due to the extreme bulk of the tert-butyl groups, only one conformation is possible: both tert-butyl groups must occupy equatorial positions to avoid the incredibly high energy associated with placing these large groups in axial positions.
Example 4: 1-Ethyl-4-methylcyclohexane
This molecule will also exist in a preferred conformation where both substituents occupy equatorial positions; however, the size difference between the ethyl and methyl groups might slightly favour one conformation over the other in the equilibrium mixture.
Advanced Considerations: Multiple Substituents and Complex Molecules
For molecules with multiple substituents, predicting the most stable conformation becomes more challenging. The overall energy difference depends on the cumulative effect of all 1,3-diaxial interactions. In such cases, computational methods can provide more accurate predictions. Furthermore, factors beyond 1,3-diaxial interactions, such as gauche interactions and steric interactions between neighboring substituents, can also play a role in determining conformational stability.
Conclusion
Understanding cyclohexane ring flips and conformational analysis is crucial for predicting the reactivity and properties of cyclohexane derivatives. The ability to select the most stable conformation based on the size and position of substituents and the understanding of 1,3-diaxial interactions is a fundamental skill in organic chemistry. By following the step-by-step approach outlined above, one can confidently predict the preferred conformations for a wide range of substituted cyclohexanes, laying a solid foundation for further exploration of organic stereochemistry. Remember that while A values provide valuable guidance, factors like specific steric interactions in larger molecules can necessitate more sophisticated analyses.
Latest Posts
Latest Posts
-
European And American Indian First Encounters Dbq
Mar 16, 2025
-
Covey Matrix Eight Dimensions Of Wellness
Mar 16, 2025
-
Color By Number Photosynthesis Answer Key
Mar 16, 2025
-
Label The Floors Of The Hotel
Mar 16, 2025
-
A Perfect Day For Bananafish Summary
Mar 16, 2025
Related Post
Thank you for visiting our website which covers about Select The Ring Flip For The Following Compound . We hope the information provided has been useful to you. Feel free to contact us if you have any questions or need further assistance. See you next time and don't miss to bookmark.
